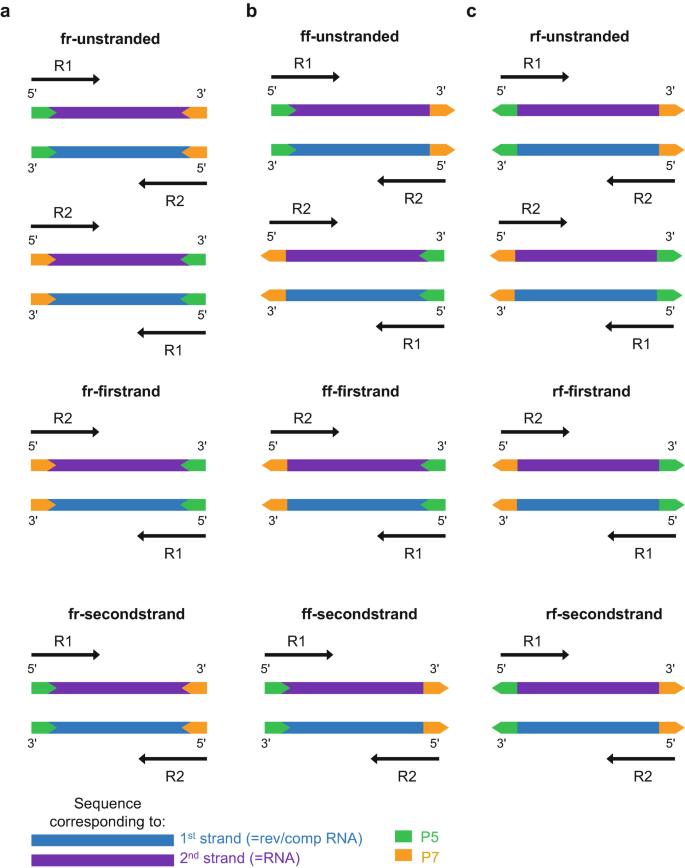
在RNA-Seq数据分析中,文库的链特异类型决定着表达定量软件的重要参数设定,除了询问构建文库的实验人员,还可以通过数据分析,得知文库类型。
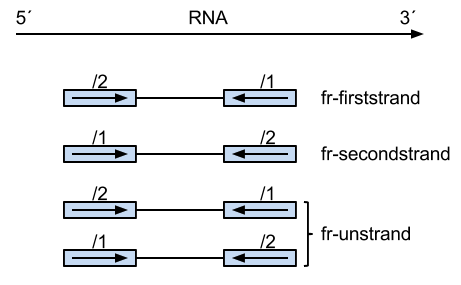
不同的链特异性类型
| 类型 | 说明 |
|---|---|
| fr-firststrand | read1的方向与基因方向相反,而read2的方向与基因一致 |
| fr-secondstrand | read1的方向与基因方向一致,而read2的方向与基因相反 |
| fr-unstrand | 非链特异性,两种情况的read都有 |
不同软件对链特异性的参数设置
| 工具 | fr-firststrand链特异 | fr-secondstrand链特异 | 非链特异 |
|---|---|---|---|
TopHat2 --library-type |
fr-firststrand |
fr-secondstrand |
fr-unstranded |
HISAT2 --rna-strandness |
R/RF |
F/FR |
NONE |
HTSeq --stranded/-s |
reverse |
yes |
no |
| Picard CollectRnaSeqMetrics STRAND_SPECIFICITY |
SECOND_READ_TRANSCRIPTION_STRAND |
FIRST_READ_TRANSCRIPTION_STRAND |
NONE |
| Kallisto quant | --rf-stranded |
--fr-stranded |
NONE |
| StringTie | --rf |
--fr |
NONE |
FeatureCounts-s |
2 |
1 |
0 |
rsem-calculate-expression--strandedness |
reverse |
forward |
none |
Salmon--libType/-l |
ISR |
ISF |
IU |
Trinity --SS_lib_type |
RF |
FR |
NONE |
| check_strandedness | RF/fr-firststrand |
FR/fr-secondstrand |
unstranded |
| RSeQC infer_experiment.py |
1+-,1-+,2++,2--占大多数 |
1++,1--,2+-,2-+占大多数 |
两者无显著差异 |
| STAR 不需要设置链特异性信息 |
NONE | NONE | NONE |
判断链特异性的方法
根据read1和read2在基因组上的比对方向,可以判断样本的文库链特异类型。
利用比对文件(bam)判断
如果已经采用STAR等不需要链特异性信息的软件完成了比对,可以直接提取比对结果进行判断。
- RSeQC软件中的infer_experiment工具可以直接读取bam进行判断
- 需要提供基因位置与链的信息(gtf/bed格式)
操作方法
安装RSeQC,并安装gtf转bed的BEDOPS软件
1 | mamba create -n BAMinfer -c bioconda bedops rseqc #利用conda安装两款软件 |
将gtf转换为bed,并通过bam文件判断链特异性reads数目:
1 | gtf2bed <genes.gtf >genes.bed #gtf转换为bed格式 |
输出:
This is PairEnd Data Fraction of reads failed to determine: 0.0087 #无法判断的reads比例 Fraction of reads explained by "1++,1--,2+-,2-+": 0.4893 #read1与基因方向一致,read2与基因方向相反的比例 Fraction of reads explained by "1+-,1-+,2++,2--": 0.5020 #read1与基因方向相反,read2与基因方向一致的比例
- 48.9%和50.2%没有显著差异,表示这是一个非链特异性文库。
- 如果
1++,1--,2+-,2-+远大于1+-,1-+,2++,2--,则为fr-secondstrand链特异 - 如果
1++,1--,2+-,2-+远小于1+-,1-+,2++,2--,则为fr-firststrand链特异
利用FASTQ文件判断
- how_are_we_stranded_here 软件可以利用fastq判断链特异性
- 软件使用kallisto将一部分reads比对到转录本,然后根据比对情况利用infer_experiment.py进行进行判读。
- 需要准备基因位置注释的gtf文件,与转录本序列文件(cDNA)
操作步骤
安装软件
1 | mamba create -n FQinfer -c bioconda how_are_we_stranded_here |
判断方向
1 | check_strandedness --gtf genes.gtf --transcripts cdna.fa --reads_1 S1.R1.fq.gz --reads_2 S1.R2.fq.gz |
输出
checking strandedness Reading reference gene model... Done Loading SAM/BAM file ... Total 20000 usable reads were sampled This is PairEnd Data Fraction of reads failed to determine: 0.0595 Fraction of reads explained by "1++,1--,2+-,2-+": 0.0073 (0.8% of explainable reads) Fraction of reads explained by "1+-,1-+,2++,2--": 0.9332 (99.2% of explainable reads) Over 90% of reads explained by "1+-,1-+,2++,2--" Data is likely RF/fr-firststrand
其他链特异类型
除了以上三种常规的fr链特异类型,根据不同的P5和P7接头方向,还有ff和rf类型,不过非常罕见。